 |
|
 |
|
|
|
Introduction
In this document we will continue to look at how the body controls cardiac output. But first a bit more on venous blood return and the supplementary pumps of the CVS.
Venous Return Mechanisms
Circulation through the blood vessels of our bodies, as you have seen, depends upon more than the power stroke (ventricular systole) of the heart.

Text and graphic courtesy Richard Klabunde. |
 |
Venous Return in Paraplegics. Previously, a student asked what happens when a person loses the ability to contract muscles in the lower body.
| Respiratory Activity (Abdominothoracic or Respiratory
Pump)
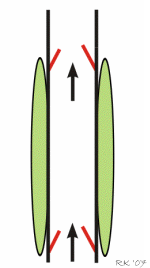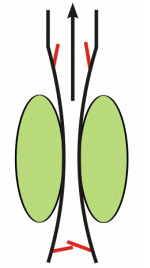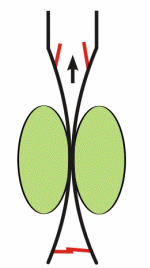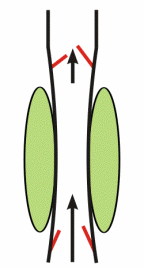
Respiratory activity influences venous return to the heart.
|
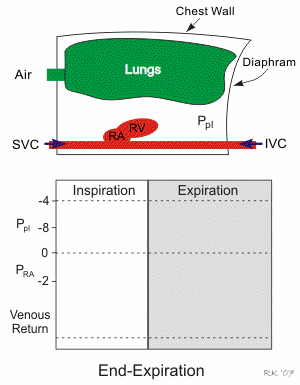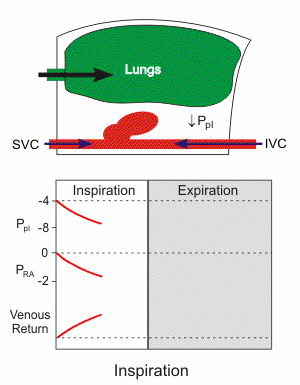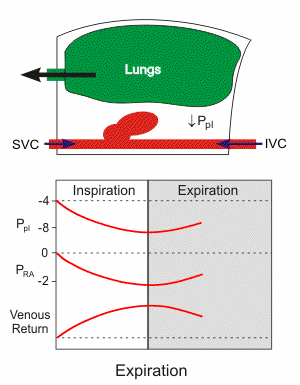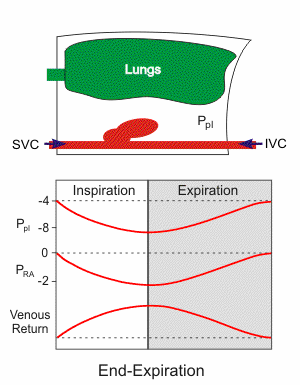 |
To summarize the above: the same muscle movement that leads to inspiration during breathing creates a negative pressure for a brief time in the right atrium of the heart. This enhances the pressure gradient between the blood of the lower extremeties and the right atrium, drawing some blood up the inferior vena cava.
Back to Cardiac Output
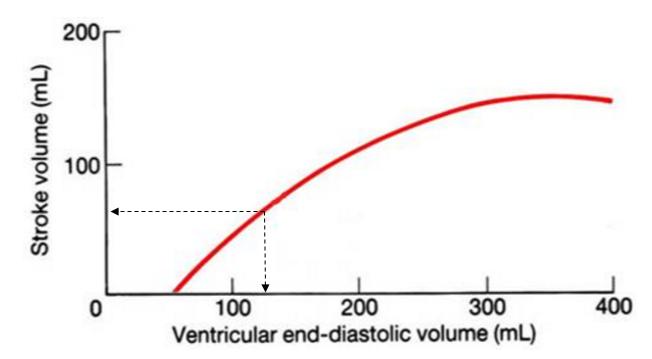
As you saw last time, the heart achieves its variable output by altering both the heart rate (HR) and the stroke volume (SV):
CO = HR * SV
The specific mechanisms
used to increase CO include:
|
|
|
|
|
 |
|
Courtesy Howard Medical School |
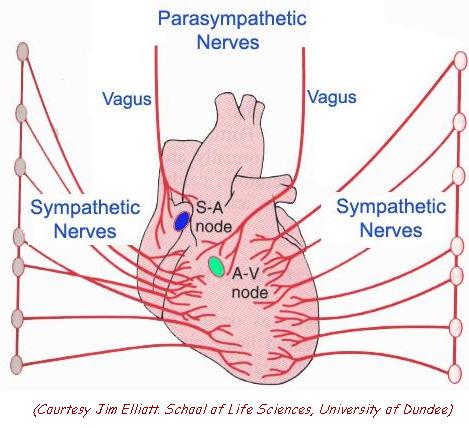
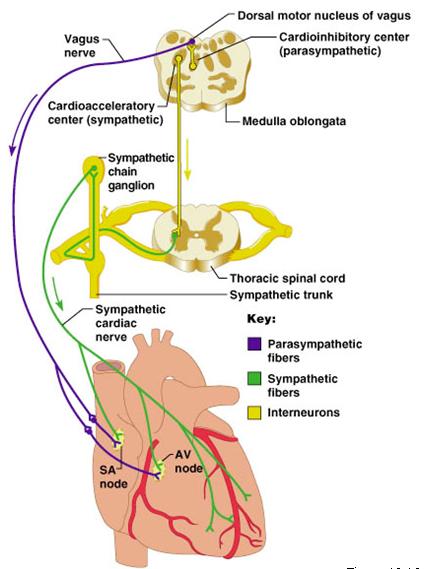
Heart Rate (HR)
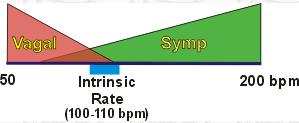
| Although resting human heart rate is ~60-80 bpm, the actual intrinsic periodicity (intrinsic rate of depolarization) of the SA node is ~100-115 depolarizations per minute. |
The first 4 figures and the following quotes courtesy of Richard E. Klabunde, Ph.D |
Before we look more closely at stroke volume, let's look at the mathematical relationship that dictates what stroke volume will be: the Bernoulli equation.
Here's the frictionless Bernoulli equation:
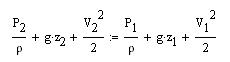
where P = pressure, z = elevation, V = velocity, and rho = blood density.
Each term in the equation has units of m2/s2.
![]()
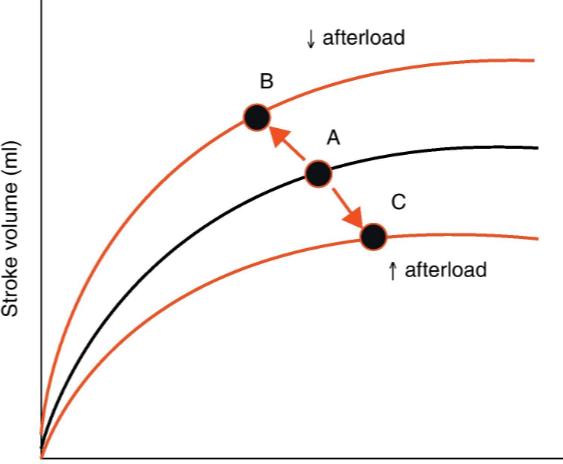
In the above equation, Pvent is affected by preload/F-S and by contractility, while Paorta (really, the P of the entire arterial tree) is affected by adjustments in afterload.
Volumtric blood flow out of the left ventricle then comes from the product of the average velocity times the cross sectional area of the opening between the ventricle and the receiving artery.
Afterload Adjustment to Compensate for Increased Cardiac Output
Changes in afterload are mainly proportional to changes in arterial cross sectional area and changes in blood velocity.
Inotropy is synonymous with contractility. Heart muscle cells are unique among muscle cells in that stimulation by the autonomic nervous system (sympathetic) can increase the speed of the myocardial responce to the arrival of a current. Skeletal and smooth muscles (the other muscle cell types found in the body) cannot do this.
If our heart rate is 60 bpm, then each complete cardiac cycle (diastole + systole) takes about 1 second. Since ventricular diastole is about 60% of the cycle, that allows about 0.6 s for the ventricle to fill and 0.4 s for ejection.
Now, let's get active: our heart rate goes up to 120 bpm. Each cycle is now only 0.5 s and filling/ejection must be accomplished in just half the time (0.3 s for relaxation + filling and 0.2 s for contraction and ejection).
Stroke Volume, Heart Rate, and Cardiac Output
Definitions of Activity
Levels. In a mathematical model created by graduate student Bruce Johnson,
(a more sophisticated version of the cardiac model you did in biosimulation),
activity levels are divided into a number of categories. These are summarized
below for an average young male.
| Level (whole body) | Description | Power Use (W) |
| Rest | Basal metabolic rate | 80 |
| Light | Can be performed indefinitely with ease | 200 |
| Moderate | Can be performed indefinitely with some effort | 350 |
| Heavy | Can be performed for 30-60 min before a break | 500 |
| Maximum | Can be performed for about 15 min before a break | 700 |
These activity levels are used in the figures, below.
Activity, Conditioning,
and Cardiac Output. The ABE CVS model was used to generate the stroke
volume, heart rate, and cardiac output predictions shown below.
|
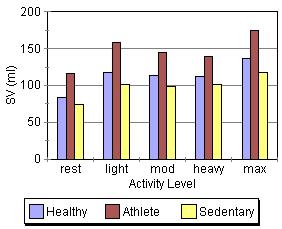
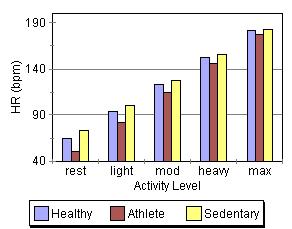
These graphs, at first glance, seem to contradict some of the things mentioned above (If changes in preload/F-S, contractility, and afterload change stroke volume, why are the stroke volume graphs nearly flat across all activity levels while cardiac output and heart rate show a steay increase?). |
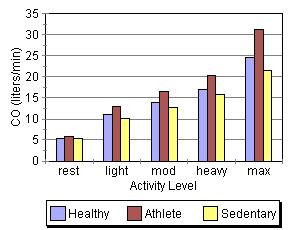 |
This apparent paradox is resolved when you recall that the time available for filling and ejection is greatly reduced as heart rate increases. The effect of preload/F-S, contractility, and afterload changes is precisely why we can hold SV constant (or show a slight increase) even though the time available for systole and diastole is reduced by about two thirds across the range of the graphs. This is quite a remarkable and accounts for how a tripling of HR can result in a 500% increase in cardiac output!
Another interesting thing shown in the graphs is the role of conditioning in cardiac performance. Athletes show the greatest increase in cardiac output despite having the lowest heart rates. Much of their superioer perfomance is due to larger stroke volumes, which are an artifact of conditioning.
The Pressure-Volume
Relationship
| You have seen that left ventricle pressure and volume can be plotted against time. | 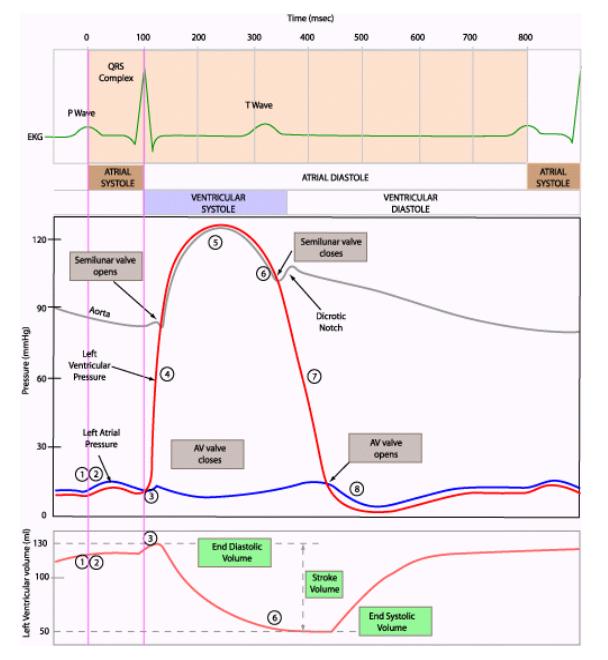 |
It is also useful and
very instructive to plot them against each other.
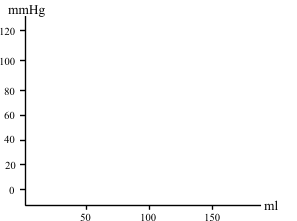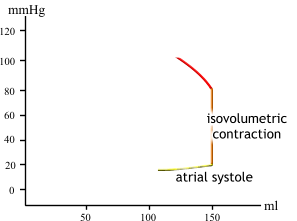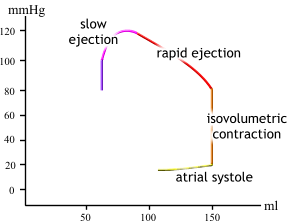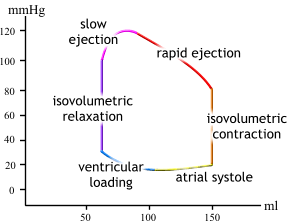
| If we do, we get a loop that just keeps going around and around. | 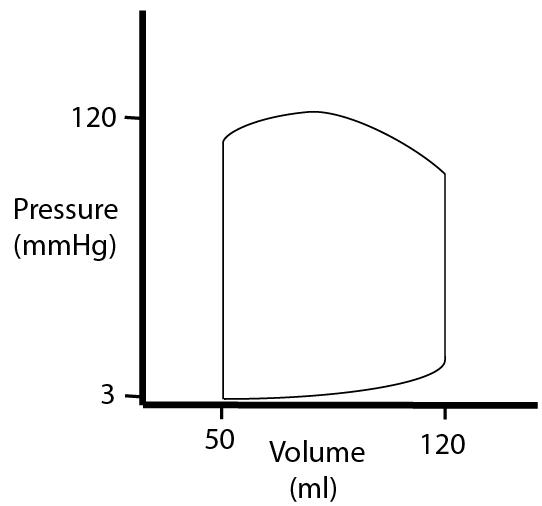 |
| During the filling part of diastole, we see the left ventricle filling at low pressure. As volume increases, the compliance of the heart wall causes pressure to increase. | 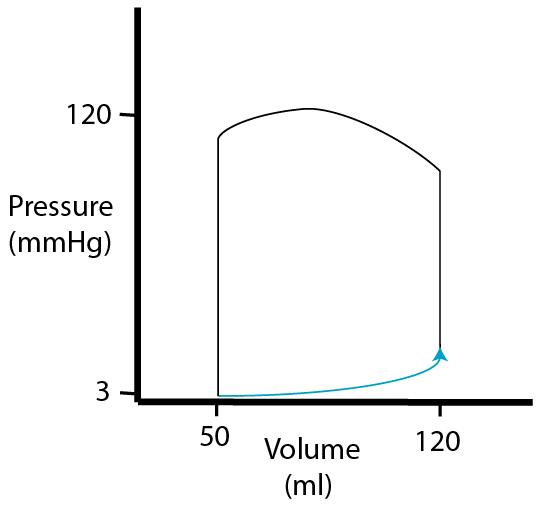 |
| At the beginning of ventricular systole, pressure increases with no change in volume (hence the vertical line, isovolumic contraction). Why does volume remain constant? | 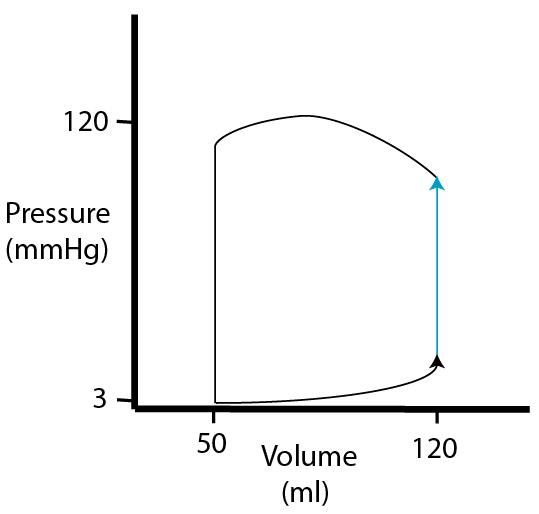 |
| During the ejection phase of systole, the tension of the ventricular muscle cells first continues to increase and then remains constant (pressure decreases because volume decreases). | 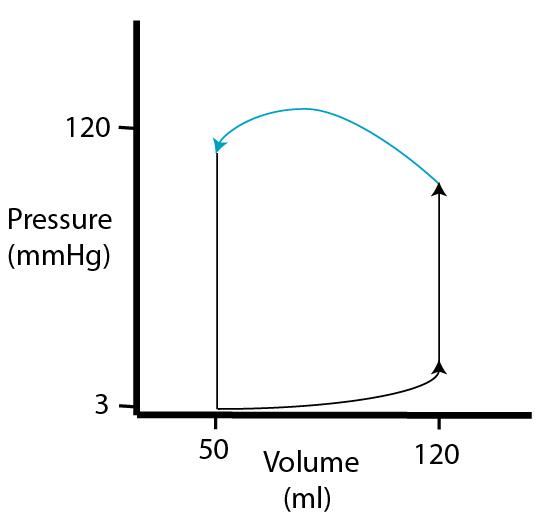 |
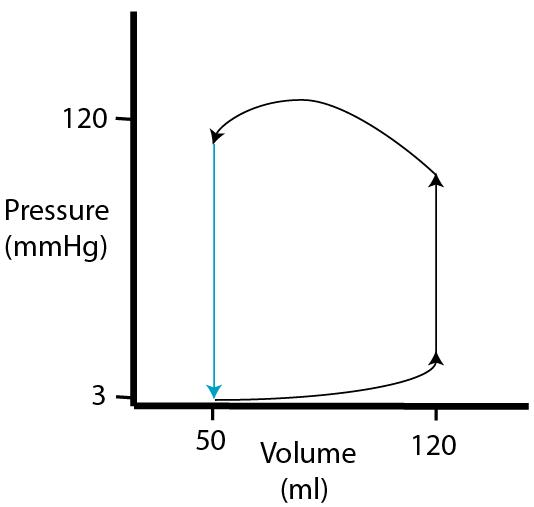
| Finally, during the
first sub
phase of diastole, pressure drops with no change in volume (isovolumic relaxation). |
 |
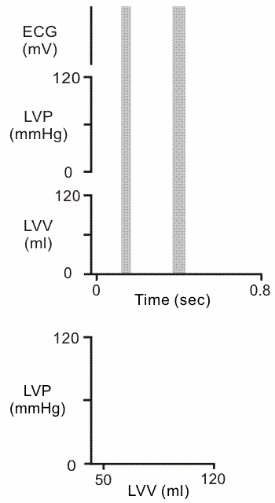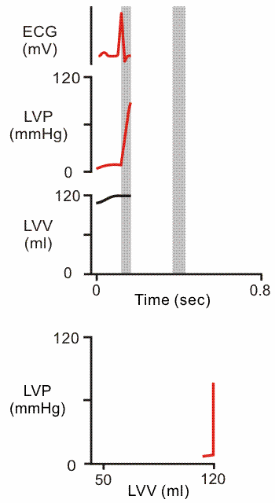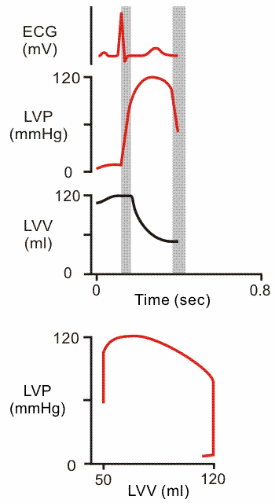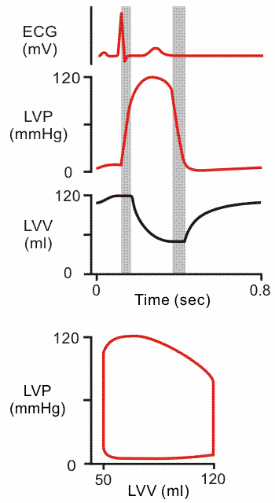
| As mentioned, stroke volume is EDV minus ESV. Stroke volume and the P-V loop will remain constant as long as activity level remains unchanged. | 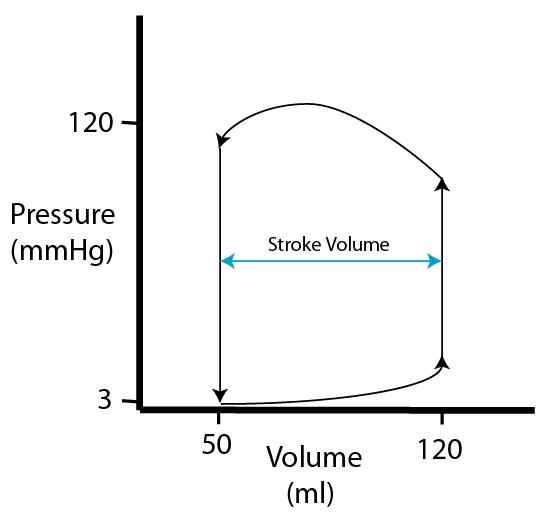 |
Some More Detail About these Plots
So now we have a loop
that goes around and around as a ventricle undergoes systole and diastole.
Let's look more closely at the shape of the plots.
| In the figure at right, "EDPVR" stands for "end diastolic pressure-volume relationship. "ESPVR" stands for "end systolic pressure-volume relationship. Let's start with EDPVR. | Image 6
|
The Bottom of the
PV Loop (EDPVR)
| You've seen the first graph at right before. It's compliance (for veins and arteries). If we reverse the axes, we have V on the x-axis and P on the y-axis (as in the P-V plots). Laid out this way (with the axes reversed), this relationship is called "elastance." Elastance is the inverse of compliance. | Image 7
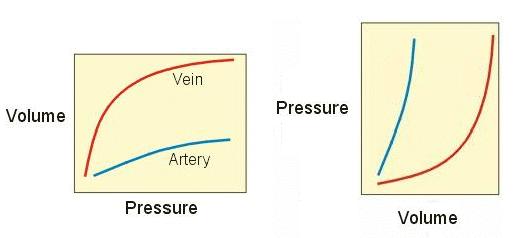 |
Notice the shape of the elastance curve (especially the red one). Does it remind you of the EDPVR line? That's because they show a similar property in both blood vessels and ventricles: as you fill them up, pressure increases. The pressure change, in both cases, is a lot like the pressure change that you see when you blow up a balloon: it's due solely to the stretching of the walls (there is no active contraction going on).
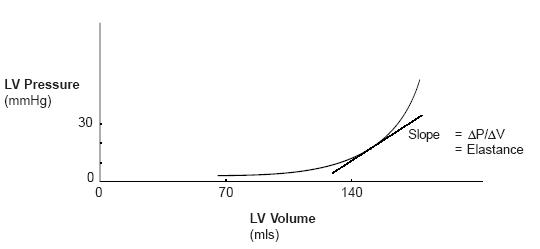
Further, the amount that pressure increases depends upon where you are in the plot.
What is the significance
of this plot?
| The graph at right shows ventricular elastance. Notice that the graph shows only a modest rise in pressure at lower volumes (dP/dV is small). Only when volumes become larger do we see that the graph turns upward (dP/dV increases). | Image 8
|
The significance of
this to the heart is two-fold.
| First, recall that the Frank-Starling mechanism results in increased force of contraction as myocardia are stretched during filling: the greater the stretch, the greater the force of contraction. But this relationship does not go on forever. It is possible to over-stretch the myocardia. If that were to happen, increased stretch would decrease force. In the graph at right, this is illustrated by the turn down in tension when myocardia are over-stretched. | Image 10
|
| In a healthy heart, this does not happen. The increase in elastance at the far right of the curve becomes a barrier that prevents over-stretching. | Image 9
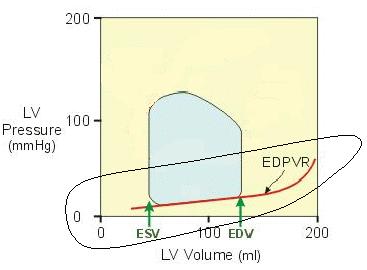 |
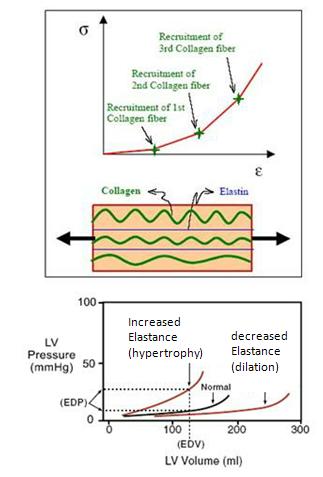
A second significant aspect of this curve is that increased elastance (hypertrophy, increased dP/dV) usually accompanies aging and can occur in certain heart pathologies. This causes the EDPVR curve to shift left, making the heart harder to fill. This is one cause of the decreased cardiac output in older people and those experiencing certain types of heart disease.
Why does stretching
of the heart wall have a characteristic curved shape?
There's no magic involved.
The upward curve in EDPVR is due to collagen and elastin in the heart walls):
|
Courtesy Technical University of Denmark |
| So now we understand
where the bottom of the PV loop comes from. It tracks the ventricular elastance
curve.
But where does the curve start? Does it start at the origin? No, it does not start at the origin. |
Image 11
|
Starting the elastance curve at the origin would suggest that adding blood to an empty ventricle would result in a pressure increase. This probably would not occur. Rather, we would expect a measurable increase in pressure with filling to occur somewhat to the right of the origin. We call this point Vo ("unstressed volume") in a PV plot. You will see its significance later.
The Top of the PV
Loop
| The vertical sides
of the loop make sense because we know about isovolumic contraction (line
BC) and isovolumic relaxation (DA).
Why does the top of the loop look as it does? Answer: The shape of the top of the loop looks as it does because the peak tension of the myocardia (the point where all the muscle cells are fully contracted) occurs between C and D.
|
Image 12
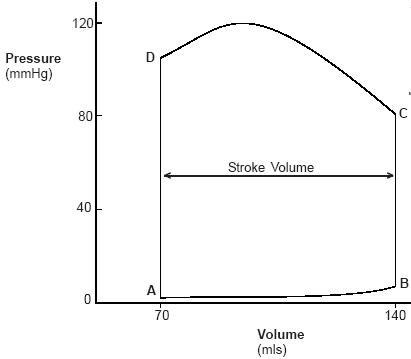 |
The Usefulness of PV loops
We've gone to quite a bit of trouble to generate and understand P-V loops. A logical question is why bother? It turns out that P-V loops are almost the only way to observe and understand how the Frank-Starling mechanism, and changes in contractility and afterload effect cardiac performance and behavior. They are also good tools for identifying and understanding a variety of heart pathologies. More on this in the next class.
A bit more on heart rate changes and reinnervation of transplanted hearts (this is FYI only)
"Heart transplant recipients can exercise and are encouraged to exercise to improve the function of the heart and to avoid weight gain. However, due to changes in the heart related to the transplant, patients should speak to their doctor or cardiac rehabilitation specialist before beginning an exercise program. Because the nerves leading to the heart are cut during the operation, the transplanted heart beats faster (about 100 to 110 beats per minute) than the normal heart (70 beats per minute). The new heart also responds more slowly to exercise and doesn't increase its rate as quickly as before." MedScape Today
We can conclude from the above that parasympathetic reinnervation is not expected to occur. There appears to be good evidence for at least some sympathetic reinnervation and perhaps, in some cases, some parasympathetic reinnervation (but perhaps not connected to the SA node): "After heart transplant (HTX), the heart is completely denervated. While sympathetic reinnervation is likely to occur, there is conflicting evidence regarding parasympathetic reinnervation" Echocardiography, Volume 23 Issue 5 Page 383-387, May 2006
"Chronotropic incompetence [i.e., the inability to change heart rate] is considered to be the main limiting factor of the functional capacity of heart transplant recipients." J Am Coll Cardiol, 1999; 33:192-197
"The reduction in
peak oxygen consumption, particularly during the first 2 years, appears
to be related in part to chronotropic incompetence. Late after transplantation
the heart rate response to exercise is greater and the decline in heart
rate after exercise faster, suggesting possible autonomic reinnervation
in some patients." J Heart Lung Transplant. 1996 Nov;15(11):1075-83.
| Finally, from the book Exercise and Sports Cardiology (published 2000): |  |
What can we conclude
from the above? For most heart transplant patients, reinnervation is probably
quite limited and CO increases are mainly due to the F-S mechanism and
perhaps to reduction in peripheral resistance (afterload). Some reinnervation
cannot be ruled out, however, suggesting that inotropy (rate of contraction)
changes may also be possible, particularly some time after the transplant.
.